SingHealth Institutions will NEVER ask you to transfer money over a call. If in doubt, call the 24/7 ScamShield helpline at 1799, or visit the ScamShield website at www.scamshield.gov.sg.
Faulty signalling pathway linked to congenital heart condition
17 Oct 2019 | Tomorrow's Medicine
By Duke-NUS
Faulty signalling pathway causes the heart to develop unnaturally while in the embryo stage, according to Duke-NUS Medical School researchers.
A congenital heart condition known as left ventricular non-compaction (LVNC) – which occurs when muscly projections in the embryonic heart fail to transform into compact heart muscle – could be caused by signalling defects, according to new preclinical research led by Duke-NUS Medical School in Singapore. The finding, published in the journal JCI Insight, could pave the way towards potential diagnosis and therapies.
During normal embryonic heart development, muscle cells migrate and proliferate, forming finger-like protrusions inside the cardiac chamber. The projections, called trabeculae, help with heart nutrition and oxygenation until blood vessels form. They eventually dissolve, leading to the formation of a compact, smooth and solid heart wall. In some embryos, however, the trabeculae fail to dissolve and form solid heart wall, leading to LVNC, in which the trabeculae project into a thinned lower left heart chamber.
“LVNC is one of the most common disorders in cardiac chamber maturation. People born with this condition often do not experience any symptoms, but can be at higher risk of heart failure, irregular heart rhythms, or sudden cardiac arrest,” said Assistant Professor Manvendra Singh, from Duke-NUS’ Cardiovascular and Metabolic Disorders (CVMD) Programme, the senior and corresponding author of the study.
Globally, scientists’ understanding of the molecular and genetic changes that lead to LVNC is limited. Asst Prof Singh and his colleagues in Singapore, Japan, and the USA wanted to test if a particular protein, Sema3E, is linked to the defects seen in LVNC. Sema3E is known to connect with a cell receptor, called PlexinD1, which is involved in embryonic heart development. However, Sema3E’s existence in the heart had not yet been determined.
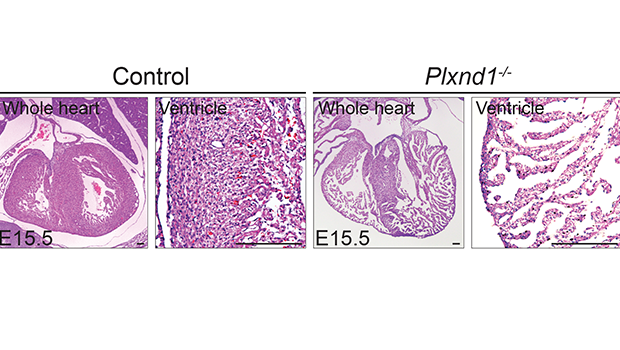
The team first turned off Plxnd1, the gene that codes for the PlexinD1 receptor, in a preclinical model of embryo hearts. This led to excessive formation of trabeculae that fail to become compacted, which typically happens in healthy heart walls.
Molecular analyses showed that turning off Plxnd1 led to increased activity from other genes initiated by a ‘Notch signalling pathway’. Inhibiting this particular pathway partially prevented the hearts from developing malformations. The team also confirmed that many different types of cells in the developing hearts expressed Sema3E. Turning off the gene that codes for Sema3E significantly reduced heart wall thickness.
“Our study helped us to better understand the role of Semaphorin-Plexin signalling in this particular congenital cardiac condition,” said Ms Reddemma Sandireddy, the study’s lead author, who is a Research Assistant in the CVMD Programme. “The results indicate that Sema3E connects with its PlexinD1 receptor to turn off the Notch signalling pathway, thus suggesting its role in the formation and maturation of the lower heart chambers.”
Professor Patrick Casey, Senior Vice Dean for Research at Duke-NUS Medical School, commented, “Currently, every third death in Singapore is due to cardiovascular diseases. Cardiomyopathy (heart muscle disease) is one of the most serious of the cardiovascular diseases, and this research expands our knowledge of one particular form of cardiomyopathy and may provide potential new targets to treat the condition.”
The researchers say further studies could reveal more information about the processes that regulate heart development and lead to therapies for this and other congenital heart conditions.
Reference: Sandireddy, R., Cibi, D., Gupta, P., Singh, A., Tee, N., & Uemura, A. et al. (2019). Semaphorin 3E/PlexinD1 signaling is required for cardiac ventricular compaction. JCI Insight, 4(16). doi: 10.1172/jci.insight.125908